hmm_sequences_dict = utils.get_filtered_dict(hmm_sequences_dict, "gene_name", set(self.default_scgs_for_taxonomy))
if not len(hmm_sequences_dict):
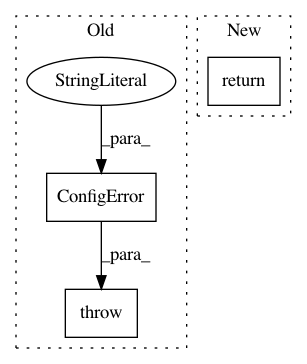
raise ConfigError("Your selections returned an empty list of genes to work with :/")
self.run.info("Num relevant SCGs in contigs db", "%s" % (pp(len(hmm_sequences_dict))))
scg_sequences_dict = {}
After Change
hmm_sequences_dict = utils.get_filtered_dict(hmm_sequences_dict, "gene_name", set(self.default_scgs_for_taxonomy))
if not len(hmm_sequences_dict):
return None
self.run.info("Num relevant SCGs in contigs db", "%s" % (pp(len(hmm_sequences_dict))))
scg_sequences_dict = {}